|
Discusión del
caso clínico
Dra. María Alejandra
Izzicupo
Se discute el
caso clínico de un paciente que consulta por diplopía,
debilidad muscular proximal, disglucia y voz nasal,
etilista y ex fumador, que presenta en el examen físico
ptosis palpebral y disminución de la fuerza de cintura
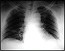
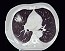
escapular y pelviana. Constatándose en la tomografía de
tórax una lesión nodular.
Vamos a tomar como dato guía la debilidad muscular como
signo que nos acerque al diagnóstico de la patología que
padece nuestro paciente.
En primera instancia, debemos tratar de localizar el
sitio de la lesión motora en la vía motora. Es decir,
según las características clínicas puede corresponder a
lesión de la motoneurona superior o inferior, el nervio
periférico, la unión neuromuscular o el músculo.
De este modo vamos a referirnos a las patologías que
pueden afectar a nuestro paciente y que fundamentalmente
pueden localizarse a nivel de la motoneurona inferior,
la unión neuromuscular y el músculo.
La esclerosis lateral amiotrófica es un trastorno
neurodegenerativo idiopático que causa fundamentalmente
debilidad muscular. Existen formas esporádicas (90%) y
familiares (10%, relacionados con mutaciones genéticas).
Se caracteriza por la degeneración de la neurona motora
y muerte con gliosis astrocítica que reemplaza la
pérdida de las neuronas. Puede manifestarse con
espasticidad, hiperreflexia, Babinski, debilidad y
atrofia muscular, fasciculaciones, calambres, disartria,
disfagia, disnea, fatiga y no presenta alteraciones
sensitivas. El diagnóstico es clínico, por
electromiografía y neuroimágenes. Los tratamientos
disponibles son poco efectivos y el pronóstico es malo.
Si bien este diagnóstico queda alejado, ya que nuestro
paciente no presenta signos de afección de motoneurona
superior, debemos tenerlo en cuenta como diagnóstico
diferencial en primera instancia y si el paciente
evolucionara desfavorablemente agregando sintomatología.
La miopatía por drogas presenta una incidencia
desconocida, pero es una causa frecuente de debilidad
muscular asociada a compromiso del músculo. Vamos a
referirnos a la miopatía por alcohol, que puede ser
aguda o crónica.
La miopatía aguda se presenta luego de la ingesta severa
con elevaciones de CPK asintomáticas hasta rabdomiólisis,
se produce por efecto citotóxico directo y se manifiesta
con calambres y tumefacción muscular. La recuperación
completa se da luego de días a semanas de suspender el
hábito.
De otro modo, la miopatía crónica es de comienzo
gradual, durante semanas a meses, con debilidad muscular
proximal sin mialgias, elevación de enzimas, ni falla
renal y la biopsia es indistinguible de otras formas de
miopatía crónica.
Otras drogas que pueden causar miopatía son:
corticoides, estatinas, fibratos, AINEs, penicilina,
sulfonamidas, cocaína, amiodarona, hidroxicloroquina,
clorpromazina, colchicina, metimazol, propiltiouracilo,
zidovudina, lamivudina, otras.
Si bien nuestro paciente refiere una ingesta de alcohol
diaria que podría estar relacionada con una miopatía
crónica por alcohol, creo que este es un diagnóstico
alejado que no puede explicar toda la sintomatología del
paciente.
La polimiositis es una miopatía inflamatoria idiomática
que se asocia a neoplasias (cáncer de mama, pulmón,
ovario, estómago y linfoma no Hodgkin) en el 9-15% de
los casos.
Sus criterios diagnósticos son: debilidad muscular
proximal simétrica, rash típico (en la dermatomiositis),
aumento sérico de enzimas musculares, alteraciones
miopáticas en la electromiografía y alteración en la
biopsia muscular.
Las manifestaciones clínicas pueden ser: debilidad
muscular proximal simétrica, atrofia muscular (en fases
avanzadas o severas), disfagia, regurgitación nasal o
aspiración, por debilidad orofaríngea o del tercio
superior del esófago, mialgias y también puede
presentarse con fiebre, pérdida de peso, poliartritis
inflamatoria no erosiva, fenómeno de Raynaud,
miocarditis y enfermedad pulmonar intersticial.
El diagnóstico se realiza por el hallazgo de los
anticuerpos (Anti-Jo1, Anti SRP, Anti-Mi2), la
electromiografía, y la biopsia muscular.
Si bien nos restan estudios diagnósticos
complementarios, creo que podemos descartar esta
entidad.
Por último vamos a referirnos a las afecciones de la
unión neuromuscular, el síndrome de Eaton Lambert y la
miastenia gravis.
El síndrome de Eaton Lambert se produce por
autoanticuerpos contra los canales de calcio
presinápticos de la placa motora, con liberación
insuficiente de acetilcolina, pero la estimulación
repetida aumenta el ingreso de calcio, liberando
suficiente acetilcolina como para desencadenar un
potencial post-sináptico. Por ello la sintomatología
mejora con el ejercicio. Es diez veces menos común que
la miastenia gravis, y en el 60% de los casos se asocia
a carcinoma de células pequeñas de pulmón (3% de éstos
pueden presentar alteraciones neurológicas que preceden
en años al diagnóstico). También puede asociarse a
enfermedades autoinmunes cuando no se asocia a
neoplasias.
Las manifestaciones clínicas son: debilidad muscular
proximal de miembros inferiores y luego superiores,
ataxia. Las manifestaciones respiratorias, bulbares y
oculares son inusuales y los síntomas autonómicos (boca
seca, constipación, impotencia, y urgencia miccional)
pueden presentarse.
El diagnóstico se realiza por la presencia de
anticuerpos anti-canales de calcio voltaje dependientes
(anti-VGCC) que son positivos en 90% de los casos y por
la electromiografía que muestra una disminución del
potencial de acción motor con incremento drástico
después de una contracción voluntaria máxima.
Creo que nuestro paciente no presenta manifestaciones
clínicas compatibles con esta entidad ya que su
debilidad aumenta y no mejora con el ejercicio repetido,
si bien queda pendiente el resultado de la
electromiografía.
La miastenia gravis (MG) es un desorden autoinmune
caracterizado por la producción de auto-anticuerpos
contra el receptor nicotínico de acetilcolina en la
membrana postsináptica de la unión neuromuscular. Puede
clasificarse:
Según edad de comienzo: - transmisión neonatal
- adulto (30 años, mujeres; 60-70años, hombres).
Según la presencia Anti.AChR: - seropositivo
- seronegativo
Según la etiología: - autoinmune
- neonatal
- drogas (D-penicilamina, curare, aminoglicósidos,
quininas, y bloqueantes cálcicos).
Según la severidad clínica:
I- Debilidad para la oclusión ocular.
II- Debilidad leve de músculos no oculares +/- debilidad
de músculos oculares con cualquier severidad.
III- Debilidad moderada de músculos no oculares +/-
debilidad de músculos oculares.
IV- Debilidad severa de músculos no oculares +/-
debilidad de músculos oculares.
V- Intubación, con ARM o no.
a- Predominio en miembros o axial.
b- Predominio orofaríngeo +/- compromiso respiratorio.
Las manifestaciones clínicas son:
Debilidad y fatiga muscular que se presenta con
actividades repetidas o sostenidas que mejora luego del
descanso, variable, inclusive durante el día. Los
factores que aumentan la debilidad pueden ser
ejercicios, altas temperaturas, infecciones, trastornos
emocionales, drogas, cirugías, embarazos.
Los músculos más frecuentemente afectados son el
elevador párpado superior, los extraoculares, los
músculos proximales de los miembros, músculos de
expresión facial, y los músculos extensores del cuello.
La ptosis palpebral mejora con el descanso o con
aplicación hielo local. Puede presentarse con dificultad
para la oclusión ocular, siendo las parálisis oculares a
menudo asimétricas y fluctuantes. La facie se observa
sin expresión, la boca puede estar abierta y el paciente
debe sujetar la mandíbula con sus dedos y cuando el
paciente logra sonreír parece que gruñe. También puede
presentar voz nasal, regurgitación nasal, disfonía,
disfagia, disnea. La progresión de la debilidad en MG
usualmente ocurre en dirección craneocaudal, los
reflejos osteotendinosos son normales o aumentados y no
se encuentran trastornos sensitivos.
El diagnóstico puede realizarse por distintas
metodologías:
El test de Edrofonio se realiza mediante la infusión
endovenosa de este inhibidor de la acetilcolinesterasa.
Su acción comienza en 30 segundos, y dura pocos minutos.
Puede presentar falsos positivos (ELA, poliomielitis,
neuropatías periféricas, lesiones troncoencefálicas,
denervación oculomotora, oftalmoplejia crónica externa,
miopatía mitocondrial, personas sanas). Debe realizarse
con monitoreo cardiaco por el riesgo de bradicardia e
hipotensión arterial.
Los anticuerpos anti-receptor de acetilcolina (Ac.
anti-AChR) se detectan por radioinmunoensayo y presentan
alta especificidad. Es positivo en el 80-85% y en el
50-60% de las formas generalizadas y oculares
respectivamente. No se correlacionan con la severidad de
la enfermedad, si bien la disminución del título de
anticuerpos puede estar relacionada con la respuesta al
tratamiento.
Los anticuerpos Anti-Musk se presentan en el 10-20% de
los pacientes y se hallan en las formas seronegativas de
la MG. La región blanco de estos anticuerpos se halla en
la unión neuromuscular es una proteína llamada “muscle
specific protein kinasa” o Musk. No se hallan con Ac.
Anti-AChR en forma conjunta.
Los tests electrofisiológicos son:
El test de estimulación nerviosa repetitiva muestra una
progresiva reducción en la amplitud del componente
muscular del potencial de acción cuando el nervio esta
sujeto a una estimulación repetitiva supramáxima a una
frecuencia de 3Hz y es positiva cuando esta reducción es
mayor al 10%. Deben testearse varios músculos o los que
presentan debilidad. Es casi siempre positivo en MG
generalizada, pero sólo es positivo en 50% de MG ocular.
La sensibilidad es del 75%.
La electromiografía de fibra única presenta una
sensibilidad mayor al 95% en MG. Mide el intervalo entre
la despolarización de cada fibra de una unidad motora y
está aumentado patológicamente en la MG.
El tratamiento puede realizarse con distintos fármacos:
Los inhibidores de la acetilcolinesterasa, aumentan la
acetilcolina disponible para actuar con el receptor. Son
las drogas de primera elección y pueden ser las únicas
utilizadas en la MG leve. No modifican el curso de la
enfermedad brindando mejoría sintomática. Se utiliza
piridostigmina (30 mg, 3 veces/día hasta 60-90 mg, 4
veces/día, según respuesta o tolerancia), presenta un
rápido comienzo de acción (15-30´), y la duración efecto
es de 4 hs. Generalmente provocan una remisión parcial
de los síntomas, y los efectos disminuyen a lo largo del
tratamiento. Como efecto adverso pueden presentar una
crisis colinérgica.
Los corticoesteroides son utilizados en la MG moderada a
severa o leve sin respuesta a inhibidores de la
acetilcolinesterasa. Se utiliza prednisona 10-20 mg/día
hasta 60 mg/día en forma progresiva y el inicio de la
respuesta se espera en 2-4 semanas (máxima respuesta
6-12 meses).
Los inmunosupresores (azatioprina, ciclofosfamida,
ciclosporina, metotrexate, micofenolato) presentan
inicio de respuesta en semanas o meses y deben comenzar
precozmente asociado a corticoides.
La plasmaféresis brinda una mejoría rápida y
transitoria, al disminuir niveles de Ac-AChR pero solo
se utiliza en MG severa o crisis miasténica.
Las inmunoglobulinas se utiliza en MG severa, crisis
miasténica o MG intratable.
La timectomía se realiza en tumores tímicos asociados o
en la MG per se observándose mejoría sintomática en el
80-85% de los casos.
Para tener en cuenta debemos recordar otras enfermedades
asociadas a MG: hiperplasia tímica, hipo-hipertiroidismo
(13%), artritis reumatoidea, anemia perniciosa, lupus
eritematoso sistémico, sarcoidosis , síndrome Sjögren,
polimiositis, colitis ulcerosa, pénfigo.
Y también aunque son pocos los casos reportados debemos
tener en cuenta los síndromes paraneoplásicos que son
trastornos de la función del sistema nervioso que
resultan del daño a este nivel sin compromiso directo
del cáncer o sus metástasis. Si bien la etiopatogenia es
desconocida se asocia a factores inmunológicos y los
anticuerpos pueden hallarse en suero y en LCR.
El compromiso muscular paraneoplásico se puede observar
en la miastenia gravis, el síndrome miasténico de Eaton
Lambert, la polimiositis/dermatomiositis entre los más
frecuentes.
En este sentido la MG paraneoplásica se ha descripsto
asociada a cáncer de páncreas, timo, carcinoma de
células pequeñas de pulmón, carcinoma neuroendócrino de
pulmón, cáncer de tiroides, mama, riñón, enfermedad de
Castleman, y enfermedad de Hodgkin.
Creo que el diagnóstico más probable para tener en
cuenta en nuestro paciente es la MG paraneoplásica,
quedan pendientes la electromiografía, los Ac. AChR, y
la biopsia pulmonar que nos ayudaran a confirmar el
diagnóstico.
Bibliografía:
1. Evaluation of the Patient with Muscle Weakness AARON
SAGUIL, CPT (P), MC, USA, Madigan Army Medical Center,
Tacoma, Washington, April 1, 2005, Volume 71, Number 7
American Family Physician.
2. Update on myastenia gravis, B R Thanvi, Postgrad.
Med. J. 2004;80;690-700.
3. Small-cell lung cancer, paraneoplastic cerebellar
degeneration and the Lambert–Eaton myasthenic
4. Síndrome, W. P. Mason,1 F. Graus,4 B. Lang,5 J.
Honnorat,6 J.-Y. Delattre,7 F. Valldeoriola,4 J. C.
Antoine,8 M. K. Rosenblum,2 M. R. Rosenfeld,1,3 J.
Newsom-Davis,5 J. B. Posner1,3 and J. Dalmau 1,3, Brain
(1997), 120, 1279–1300.
5. Clinical correlates with anti-MuSK antibodies in
generalized seronegative myasthenia gravis ,Amelia
Evoli,1 Pietro A. Tonali,1 Luca Padua,1 Mauro Lo
Monaco,1 Flavia Scuderi,2, Anna P. Batocchi,1 Mariapaola
Marino2 and Emanuela Bartoccioni2, Brain (2003), 126,
2304±2311
6. Pancreatic Cancer in a Patient with Myasthenia Gravis
ROSALIA PATTI, MD,* ERALDO NATAL` E, MD, and ALBERTO
MARINGHINI, MD, Digestive Diseases and Sciences, Vol.
49, No. 6 (June 2004), pp. 990–991 ( C° 2004)
7. Myasthenia gravis as a paraneoplastic syndrome
associated with renal cell carcinoma Erik l. Torgerson,
Ramin Khalili, From the Department of Urology, The
journal of urology vol. 162, 154, july 1999
8. Castleman’s Disease Associated With Myasthenia Gravis
Jonathan R. S. Day, MRCS, Duncan Bew, MBBS, Mark Ali,
FRCS, Roberto Dina, FRC(Path), and Peter L. C. Smith,
FRCS, Ann Thorac Surg
9. 2003;75:1648–50.
10. Evaluating muscle symptoms, Richard Petty J. Neurol.
Neurosurg. Psychiatry 2003;74;38-42
11. Myasthenia gravis. Drachman, DB. N Engl J Med 1994;
330:1797.
12. The treatment of patients with myasthenia gravis.
Sanders, DB, Scoppetta, C. Neurol Clin 1994; 12:343.
13. Treatment of autoimmune myasthenia gravis. Richman,
DP, Agius, MA. Neur
|
|
|