Discusión del caso clínico. | Presentación |
Como objetivos de la discusión nos planteamos :
-
Describir el abordaje del paciente con debilidad aguda.
-
Repasar conceptos generales y la clasificación de las neuropatías periféricas.
-
Analizar y describir las etiologías más probables de neuropatía en nuestra paciente.
-
Revisar la relación entre neuropatías y embarazo /puerperio.
-
Realizar consideraciones finales.
El desafío inicial es determinar exactamente lo que significa un paciente cuando se queja de debilidad. “La debilidad real es la incapacidad de realizar un movimiento deseado con fuerza normal debido a una reducción en la fuerza muscular”. Aunque esto puede deberse a un proceso neuromuscular primario, más a menudo la queja representa un malestar de enfermedad médica.
La disfunción motora puede ser resultado de la afectación de la motoneurona superior o de la motoneurona inferior, así como de la unión intramuscular y las fibras musculares.
El examen de la fuerza muscular nos hará distinguir la debilidad subjetiva, en la que no hay evidencias de pérdida de fuerza; de la objetiva puesta de manifiesto en la exploración física. Así mismo, debemos distinguirla de la astenia (sensación subjetiva de falta de energía) y de la ataxia (incapacidad para realizar movimientos voluntarios de origen cerebeloso). El problema residirá, por tanto, en alguno de los eslabones de la vía de la motricidad voluntaria, desde que nace el estímulo cortical motor hasta que se produce la contracción muscular.
La historia debe incluir una descripción de la distribución de la debilidad y sus manifestaciones, el período durante el cual se desarrollaron los síntomas y características clínicas asociadas con la debilidad (p. ej., afasia, diplopía). Es importante determinar si la debilidad es unilateral o bilateral y si hay signos asociados con la afectación neurológica central.
Para una evaluación objetiva del patrón y el grado de debilidad muscular, es necesaria una exhaustiva exploración muscular. La parálisis de la motoneurona superior (lesión de la vía piramidal) produciría: espasticidad, hiperreflexia tendinosa, reflejo cutáneo plantar, sin atrofia o con modera atrofia por desuso y sin fasciculaciones. Si encontramos atrofia, flaccidez, hiporreflexia o arreflexia tendinosa y fasciculaciones o anestesias, estaremos ante una parálisis de la motoneurona inferior.
Nuestra paciente se manifestó con paraparesia , por lo tanto me planteo los diagnósticos diferenciales de esta debilidad aguda bilateral.
Ataque cerebrovascular: las lesiones en el tronco encefálico pueden producir afectación de nervios craneales ipsilaterales y debilidad del cuerpo contralateral. Los hallazgos clínicos varían dependiendo de las áreas involucradas.
Enfermedad de la médula espinal : se puede sospechar cuando hay signos o síntomas motores y sensoriales bilaterales que no afectan al cerebro. Los déficits motores implican debilidad, espasticidad muscular e hiperreflexia (aunque la hiporreflexia suele verse en agudo); los hallazgos sensoriales implican un nivel sensitivo por debajo del cual la sensación está ausente o reducida. Una amplia gama de patologías puede causar enfermedad de la médula espinal, incluyendo trauma, infección (por ejemplo, absceso epidural), neoplasia, hemorragia, inflamación (por ejemplo, mielitis transversa) y trastornos degenerativos.
Enfermedad del nervio periférico: la causa más frecuente y de mayor gravedad es el Síndrome de Guillain-Barré (GBS). Harémos referencia a esta patología más adelante.
Enfermedad de la unión neuromuscular: La Miastenia Gravis (MG) puede producir debilidad en cualquier grupo muscular. Algunas presentaciones son más comunes: síntomas oculares en el 50 % ;bulbares en el 15 % ; y la debilidad de la extremidad aislada se produce en alrededor del 5 %. El envenenamiento por organofosforado se caracteriza por manifestaciones de exceso colinérgico(bradicardia, miosis, lagrimeo, salivación, broncorrea, broncoespasmo, micción, emesis, diarrea, diaforesis y debilidad generalizada). El botulismo se manifiesta con pródromos de vómito, dolor abdominal, diarrea y sequedad de boca. Los síntomas de la afectación del nervio craneal se desarrollan seguido de debilidad muscular descendente, que suele progresar desde el tronco y las extremidades superiores hasta las extremidades inferiores.
Enfermedad del músculo: La miopatía alcohólica suele ser crónica, manifestándose con calambres musculares, disminución de la sensibilidad y edema, siendo una causa importante de rabdomiólisis no traumática. Las miositis tanto la dermatomiositis como la polimiositis suelen presentarse con una debilidad muscular proximal simétrica, que a menudo se ha venido agravando durante varios meses.
Consideramos por las manifestaciones clínicas y por los estudios complementarios la afectación en nuestra paciente corresponde a una enfermedad del nervio periférico.
Neuropatías periféricas
Son un grupo diverso de trastornos que pueden ser adquiridos o hereditarios, de origen sistémico o restringidas a los nervios periféricos.
Puede clasificarse según: inicio:agudo, subagudo, crónico; distribución anatómica: afectación de nervios craneales, miembros superiores o inferiores, músculos respiratorios, simétricos o implicación asimétrica; fibras nerviosas afectadas: sensoriales, motoras o de fibras autonómicas; patrón fisiopatológico: axonal o desmielinizante; etiología: causa subyacente de la enfermedad (trastorno inmunológico, cáncer, infección, toxinas, trastorno metabólico o enfermedad hereditaria).
Manifestaciones clínicas: Cuando los nervios motores están comprometidos, la neuropatía se manifiesta con debilidad y atrofia muscular. El daño a los nervios sensitivos puede causar pérdida de la sensibilidad, parestesias y disestesias, dolor y ataxia sensorial. El compromiso de las fibras pequeñas no mielinizadas causa la pérdida de la sensibilidad al dolor y a la temperatura, adormecimientos y parestesias dolorosas y cuando se afectan las fibras sensitivas largas puede haber pérdida de las sensaciones de vibración y posición, ataxia sensorial y parestesias tipo adormecimiento o picazón. La disfunción autonómica puede resultar en hipotensión postural, disfunción sexual, disfunción gastrointestinal y genitourinaria, sudoración anormal y pérdida del cabello. Los reflejos tendinosos profundos frecuentemente están disminuidos o ausentes, particularmente en las neuropatías desmielinizantes. Dado que la mayoría de los troncos nerviosos son mixtos, el daño a los nervios periféricos a menudo afecta más de una de estas funciones.
Diagnóstico: Los estudios electrodiagnósticos, incluyendo la electromiografía y los estudios de conducción nerviosa, distinguen las neuropatías axonales de las desmielinizantes, las cuales comúnmente son inflamatorias y tratables. La biopsia del nervio y del músculo es particularmente útil en el diagnóstico de las neuropatías autoinmunes o vasculíticas y las secundarias a sarcoidosis o por amiloide. Los estudios sanguíneos o del LCR pueden ayudar en el diagnóstico de las neuropatías inflamatorias, paraneoplásicas, infecciosas, endocrinas, metabólicas, tóxicas, nutricionales o hereditarias.
La paciente presentaba una polineuropatía simétrica aguda. Dicha entidad no es tan frecuente pero es muy importante porque su causa más común es el síndrome de Guillain-Barré, que puede ser fatal. En el siguiente cuadro se señalan otras causas:
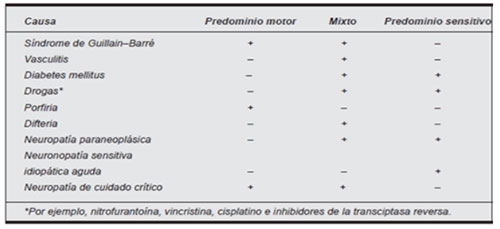
Neuropatía y diabetes
La neuropatía diabética (ND) es la más común de las complicaciones crónicas de la diabetes, afecta a más del 50% de los pacientes. Abarca un grupo de síndromes clínicos y subclínicos con diferentes manifestaciones caracterizados por daño focal o difuso del sistema nervioso somático y/o autonómico. En más de la mitad de los casos, la neuropatía es asintomática. Desde el punto de vista práctico, es fácil enfocar la neuropatía de la siguiente forma:
Neuropatía difusa: polineuropatía sensorio-motora simétrica distal; principalmente neuropatía de fibras pequeñas; neuropatía de fibra grande.
Neuropatía autonómica: función pupilar anormal; disfunción de la sudoración; neuropatía autonómica genitourinaria (vesical y sexual); neuropatía gastrointestinal autonómica (atonía gástrica, diarrea diabética); neuropatía medular adrenal; neuropatía autonómica cardiovascular; hipoglucemia.
Neuropatía focal: mononeuropatía (extremidad superior o inferior); mononeuropatía multiple; plexopatía; polirradiculopatía (puede ocurrir con neuropatía difusa); mononeuropatía craneal.
Nuestra paciente es diabética, sin embargo creo que esta no es la causa de su problema actual porque como se mencionó anteriormente dicha enfermedad se manifiesta de manera solapada, con una afectación crónica.
Neuropatías tóxicas
Las neuropatías tóxicas son relativamente infrecuentes, ocurren entre el 2 al 4% , con menos frecuencia que las de origen metabólico, hereditario. Están causadas por los siguientes factores: agentes farmacéuticos (antimicrobianos, antimicóticos, antirretrovirales, inmunosupresores) considerados los más frecuentes; agentes biológicos incluidos de animales, plantas o bacteriológicos; agentes por exposición ocupacional o medioambientales (metales pesados).
Los test toxicológicos generalmente no son útiles porque no están disponibles y porque la sustancia no es detectable debido al retraso entre la examinación y la exposición.
Descartamos esta etiología ya que nuestra paciente no presentó ningún tipo de exposición tóxica según refiere. Además, estos cuadros también se manifiestan de manera insidiosa no siendo compatible con el debut agudo de nuestro caso.
Neuropatía y porfiria
La porfiria intermitente aguda (PIA) es un trastorno autosómico dominante que resulta de una deficiencia parcial de porfobilinógenodesaminasa, la tercera enzima en la vía hemobiosintética.
La mayoría de los individuos con porfiria intermitente aguda nunca desarrollan síntomas. Por consiguiente, la enfermedad sintomática puede omitir generaciones o ser reconocida en un solo individuo dentro de una familia. La presentación es muy variable y los síntomas son inespecíficos, lo que explica en parte los retrasos en el diagnóstico. Los síntomas generalmente ocurren entre la tercera y cuarta década, en mujeres principalmente, presentándose como ataques agudos, los cuales son precipitados por tabaco, alcohol, estrés, dieta pobre en calorías y cambios hormonales cíclicos en mujeres, existiendo recaídas durante el periodo premestrual y embarazo. Las manifestaciones más comunes de PIA son gastrointestinales (dolor abdominal 85-95%), neurológicas (paresia 40-70%) y síntomas psiquiátricos (40-58%).
En cuanto a la neuropatía periférica en la porfiria , la afectación sensitiva es común y se presenta con dolor en la espalda, el pecho y las extremidades, y puede preceder al dolor abdominal. La debilidad motora generalmente comienza proximalmente en las extremidades superior e inferior y puede progresar distalmente. Especialmente con ataques prolongados, también puede involucrar nervios craneales y conducir a parálisis bulbar, deterioro respiratorio y muerte.
El diagnóstico definitivo se realiza con la demostración del exceso de ácido delta aminolevulínico, porfobilinógeno y de porfirinas totales en orina y sangre.
Consideramos poco probable dicha causa ya que la paciente no tiene antecedentes familiares de porfiria, no presentó síntomas gastrointestinales que son los más frecuentes y la afectación neurológica no fue la típica de esta enfermedad. De todos modos pensamos que hubiese sido de utilidad el dosaje de porfirinas para un adecuado algoritmo diagnóstico.
Síndrome de Guillán Barré
El síndrome de Guillain-Barré (GBS) fue descripto por primera vez en 1916 como una forma "benigna" de debilidad de la extremidad asociado a la recuperación total. Actualmente se reconoce como una polirradiculoneuropatía desmielinizante aguda, monofásica, inflamatoria, con diferentes variantes clínicas.
La incidencia oscila entre 0,40 y 3,25 casos por 100.000 personas, dependiendo sobre la localización geográfica.
La asociación con infecciones (tracto gastrointestinal y respiratoria principalmente) es bien conocida, siendo el Campylobacter jejuni el principal patógeno asociado. También se han descripto casos precipitados por virus Epstein-Barr, influenza A, Mycoplasma pneumoniae, Haemophilus influenzae, hepatitis (A, B y E) y el virus Zika. También se han descripto asociaciones con la vacuna de la gripe, cirugías y situaciones de stress, estando estas causas no bien definidas aún.
En los casos típicos (GBS clásico), el cuadro inicia con dolor, entumecimiento, parestesia o debilidad en las extremidades con afectación bilateral rápidamente progresiva con o sin compromiso de los músculos respiratorios o craneales. Los criterios diagnósticos son la debilidad motora progresiva de más de un miembro y arreflexia o hiporreflexia marcada.
Existen variantes clínicas: GBS Clásico: tetraparesia con o sin afectación motora del nervio craneal; GBS paraparético de miembros inferiores; Debilidad faríngeo-cervical-braquial: bulbar, cuello y extremidades superiores; Debilidad bifacial con parestesias: facial; Síndrome de Miller Fisher: oftalmoplejía externa; Encefalitis de Bickerstaff: oftalmoplejía externa.
El síndrome de Guillain-Barré es a menudo una enfermedad postinfecciosa, con lesiones nerviosas mediadas por el sistema inmunológico. Existen tres fenotipos: desmielinizantes, puramente axonal, y desmielinizante con compromiso axonal. La inmunopatogénesis difiere en cada uno de estos modelos conceptuales, y la respuesta al tratamiento es también diferente. La hipótesis actual de la inmunopatogénesis de SGB sostiene que este es mediado por anticuerpos. No todos los anticuerpos antigangliósidos son neurotóxicos, pero los anticuerpos gangliósidos GM1 o GD1a (en los nodos de Ranvier) activan el complemento que destruye la mielina.
El examen del LCR típicamente muestra un alto valor de proteínas con recuento de células normales (disociación albúmino-citológica).Dicha proteinorraquia suele aumentar progresivamente en el transcurso de las semanas. Sin embargo, en un 10% a 25% de los casos no existen alteraciones en el LCR.
La electromiografía puede ser útil para confirmar el diagnóstico en casos clínicamente difíciles, como en pacientes con dolor extremo, y es particularmente necesario para subclasificar GBS en los subgrupos de neuropatía axonal motora aguda (NAMA) y la polineuropatía desmielinizante inflamatoria aguda (PDIA) .
Tratamiento
Inmunoterapia: la plasmaféresis elimina anticuerpos y otros posibles componentes inflamatorios tal como el complemento y puede mejorar la función supresora de las células T. Los grandes ensayos multicéntricos han establecido la eficacia de dicho tratamiento con evidencia de mejoría clínica y reducción de la necesidad de ventilación mecánica. Alternativamente, se puede utilizar inmunoglobulina endovenosa (IGEV) a 0,4 g / kg por día de infusión a aproximadamente 1 a 3 ml / min por 5 días.
Dos grandes ensayos controlados han encontrado IGEV y plasmaféresis tienen una eficacia similar, pero la primera presentó menos complicaciones. Ambos tratamientos son más efectivos dentro de las 2 semanas de inicio de síntomas. La combinación de terapias no demostró mayor eficacia y la adición de corticoides no manifestó beneficios.
GBS y su relación con el embarazo /puerperio
La paciente presentó esta paraparesia aguda en el contexto de un post aborto inmediato, previo a la realización del legrado quirúrgico.
No hay descripta en la bibliografía aumento de la incidencia de dicha entidad durante el embarazo, sí durante el puerperio, principalmente durante las primeras dos semanas. Existen varios reportes de casos que describen dicho cuadro.
Consideraciones finales
Creo que el síndrome de Guillán Barré variante paraparética por una neuropatía axonal motora aguda es la causa más probable de la afección de nuestra paciente.
Es compatible la clínica y los hallazgos electromiográficos. El que no haya presentado la disociación álbumino –citológica alejó el diagnóstico de esta enfermedad inicialmente pero como mencioné anteriormente en un porcentaje no menor el LCR es normal.
De ser este el diagnóstico en nuestra opinión ya no estaría indicado el tratamiento específico ya que han pasado tres semanas del inicio de los síntomas y la paciente ha presentado mejoría espontáneamente.
Deberemos continuar con la rehabilitación motora y evaluación clínica interdisciplinar continua y destacamos la importancia de pensar siempre en las manifestaciones atípicas de enfermedades comunes antes que en las enfermedades raras.
Bibliografía:
1 .Jose Antonio Liranzo Medina. “Pérdida de fuerza” .Medicina Familiar y Comunitaria Servicio de Urgencias Hospital Clínico Universitario .MALAGA.
2. Andrew W Asimos,et al . “Evaluation of the adult with acute weakness in the emergency department”.Up to Date.2013.
3. John D England,et al. “ Peripheral neuropathy”. THE LANCET .Vol 363.June 26.
4. Michelle L. Mauermann; Ted M. Burns; “Pearls and Oy-sters: Evaluation of peripheral neuropathies” Neurology 2009;72;e28-e31.
5. Vital A, LarrivitrcM,et al . “Severe axonal polyneuropathy with onset inthe postpartum period”.ActaNeurolScand 1994: 89: 303-306.
6. Actualización de consenso de neuropatIa diabética.2009.Comité de Neuropatía Diabética de la Sociedad Argentina de Diabetes.
7. Fernanda Campos da Silva, Gláucio de MoraesPaula,et al. “Guillain-Barré Syndrome in Pregnancy: Early Diagnosis and Treatment Is Essential for a Favorable Outcome”. GynecolObstetInvest 2009;67:236–237.
8. Gustavo Pradilla. “ Diagnóstico y tratamiento de las neuropatías periféricas”.Cap 14.
9. Clarissa Crone ,ChristianKrarup. “Diagnosis of acute neuropathies”.JNeurol (2007) 254:1151–1169.
10. Benjamin R Wakerley,Nobuhiro Yuki. “Mimics and chameleons in Guillain–2Barré and Miller Fisher syndromes”.Wakerley BR, et al. PractNeurol 2015;15:90–99.
11. Bianca van den Berg,ChristiaanFokke,et al. “ParapareticGuillain-Barré syndrome”.2014 American Academy of Neurology.
12. Pieter A van Doorn, Liselotte Ruts. “Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome”. Lancet Neurol 2008; 7: 939–50.
13. F.M. Wijdicks,C. J. Klein,et al . “Guillain-Barré Syndrome”. March 2017;92(3):467-479.
14. Nobuhiro ,HHartung; “.Guillain–Barré Syndrome”. nengl j med 366;24 nejm.2294 org june 14, 2012.
15. GazquezS,KMavilab.“ La porfiria aguda intermitente, un problema diagnóstico”. GastroenterolHepatol. 2010; 33 (6):436–439.
16. ABNER LOZANO L. “Porfiriaaguda : reporte de caso y revisión de tema” .L. 1,106 rev.fac. med 16 (1): 106-114, 2008”.
17. A. Tracy,et al. “Porphyria and its neurologic manifestations”. Handbook of Clinical Neurology, Vol. 120 (3rd series) .Neurologic Aspects of Systemic Disease Part II.
18. Eva L Feldman,et al. “Epidemiology and classification of diabetic neuropathy”.Up to Date.2013.
19. MHinojosa. “Revisión bibliográfica: Neuropatías tóxicas”. Instituto Nacional de Ciencias Médicas y Nutrición "Salvador Zubirán".Departamento de Medicina del Dolor y Paliativa.
|
