|
Discusión del
caso clínico
Dra. Gisela Goldberg
Nos encontramos frente a un paciente varón, adulto joven, sin antecedentes de jerarquía que consulta por un cuadro de pérdida de peso y sudoración nocturna, evidenciándose al examen físico poliadenopatías, múltiples masas subcutáneas, una tumoración a nivel clavicular y paraparesia crural, presentando por resonancia magnética (RMI) una importante masa intra-extrarraquídea. En el laboratorio se objetivan anemia de enfermedades crónicas, eosinofilia y trombocitosis, VES acelerada, aumento de LDH, hiperuricemia e hiperfosfatemia.
Los interrogantes que me planteo en relación al paciente en discusión son:
- Utilizando como dato guía inicial las adenopatías, ¿Cuáles son las probables etiologías del cuadro que presenta nuestro paciente?
- ¿Las fracturas que sufrió el paciente pueden considerarse de carácter patológico? En ese caso, ¿Qué otras etiologías debemos tener en consideración?
- ¿Pueden los datos del laboratorio darnos una pista hacia el probable diagnóstico?
- Dentro de las múltiples etiologías del síndrome de compresión medular, ¿Cuál sería la más probable en nuestro paciente?
Para introducirnos en la discusión, recordemos que las adenopatías son un motivo de consulta o un hallazgo frecuente particularmente en el ámbito de atención primaria de la salud (APS). Más de las 2/3 partes de los pacientes presenta causas inespecíficas o infecciones de vías aéreas superiores, y menos de 1% padece un tumor maligno. La mayoría de los pacientes con linfadenopatías requieren pocos exámenes complementarios para arribar al diagnóstico.
Después de los 40-50 años de edad aumenta la incidencia de procesos malignos.
La lista de enfermedades capaces de provocar afectación ganglionar es muy amplia y variada.
Teniendo en cuenta la extensión del compromiso ganglionar se pueden clasificar en localizadas o generalizadas. Se consideran generalizadas si los ganglios linfáticos están aumentados en 3 o más áreas no contiguas, como es el caso de nuestro paciente. Esta clasificación puede tener un correlato etiológico, ya que las generalizadas suelen presentarse en procesos sistémicos infecciosos (en la mayoría virales), y enfermedades linfo/mieloproliferativas. 
Las adenopatías generalizadas de forma poco frecuente son manifestación de una neoplasia, pero pueden presentarse en pacientes con linfomas, leucemias o diseminación metastásica de un tumor sólido. En pacientes inmunocomprometidos o aquellos con diagnóstico de HIV deben considerarse otros diagnósticos diferenciales: tuberculosis, criptococosis, citomegalovirus, toxoplasmosis, sarcoma de Kaposi e incluso la misma primoinfección por HIV.
En la búsqueda de la etiología de las linfadenopatías el médico se debe ayudar de una anamnesis exhaustiva, del examen físico, exámenes complementarios seleccionados y evaluar la necesidad de realizar una biopsia escisional.
Anamnesis: La edad del paciente y el contexto en el cual aparecen adenopatías pueden dar la primera orientación hacia la causa. Es necesario descartar la presencia de síntomas respiratorios. Se debe indagar sobre: ocupación; hábitos (tabaco, conductas de riesgo: enfermedades de transmisión sexual, adicciones, etc.), consumo de fármacos, viajes recientes, contacto con animales; presencia de síntomas constitucionales (fiebre, sudoración, pérdida de peso, astenia).
Examen físico: Evaluar las características semiológicas de los ganglios y examinar las regiones drenadas por los mismos así como las manifestaciones que frecuentemente se asocian a adenopatías en el contexto de enfermedades sistémicas.
Se deben consignar datos como tamaño, forma, consistencia, sensibilidad, signos de inflamación, fijación a planos superficiales o profundos lo que nos puede ayudar a tener una orientación etiológica.
La presencia de hepatoesplenomegalia, exantema, artritis, fiebre y pérdida de peso puede orientar hacia enfermedades malignas, infección sistémica, colagenopatías.
Si la anamnesis y la exploración física sugieren una causa benigna de linfadenopatía, como ocurre habitualmente en el ámbito ambulatorio, no se requerirán de estudios adicionales. Estos serán necesarios aproximadamente en la mitad de los casos y finalmente la necesidad de realizar una biopsia ganglionar se presenta en menos del 5% de los casos.
Laboratorio: Si el diagnóstico es incierto se comienza con un hemograma con fórmula leucocitaria, el hepatograma (GOT, GPT, FA, bilirrubina), LDH, VES y algunos estudios serológicos.
Finalmente la necesidad de realizar una biopsia escisional debe ser evaluada de forma individual en cada paciente y según la sospecha clínica.
En relación a nuestro paciente, en primer lugar vamos a abordar las etiologías infecciosas. La linfadenitis tuberculosa es la forma de presentación extrapulmonar más frecuente, usualmente se presenta como una adenopatía aislada en un paciente joven con síntomas sistémicos, pero puede presentarse con poliadenopatías. La localización a nivel cervical representa el 63-77% de los casos.
Suele tratarse de la reactivación de una infección latente.
Los cultivos de esputo positivos son poco frecuentes en el contexto de afección ganglionar y la radiografía (Rx) de tórax no suele evidenciar lesiones de tuberculosis (TBC) pulmonar activa.
El derrame pleural secundario a una pleuritis tuberculosa usualmente es unilateral de leve a moderada cuantía, tratándose de un exudado con valores de LDH altos (>500). La Rx de tórax demuestra compromiso del parénquima asociado a la presencia de derrame en más del 50% de los pacientes.
En cuanto al compromiso óseo y articular, comprende 10-35% de los casos de TBC extrapulmonar y 2% de la totalidad de los mismos. Suele haber afectación espinal hasta en la mitad de los pacientes.
La TBC espinal (mal de Pott) afecta más frecuentemente la columna lumbar y dorsal baja, pudiendo llegar a producir un síndrome de compresión medular. El compromiso inicialmente es óseo y del disco y posteriormente puede extenderse a los tejidos blandos adyacentes.
La cutánea es una localización poco frecuente, representando tan sólo el 1% de las formas extrapulmonares. Entre las múltiples formas de presentación se encuentra la TBC colicuativa que es el proceso subcutáneo, cuya lesión elemental suele ser un nódulo o un ganglio reblandecido, el cual avena al exterior con compromiso de la piel suprayacente. Suele ocurrir en personas jóvenes (sobre todo niños).
Si bien es una patología altamente prevalente en nuestro medio, el cuadro clínico no parece coincidir con el del paciente en discusión, por lo que creo que es un diagnóstico alejado. Sin embargo, considero adecuado descartar el mismo.
Las micosis sistémicas (coccidioidomicosis, paracoccidioidomicosis, criptococosis, histoplasmosis) carecen de manifestaciones clínicas específicas y la afección subcutánea puede confundirse con infecciones similares de otro origen. Suelen afectar a pacientes inmunosuprimidos.
Se presenta con síntomas constitucionales (fiebre, pérdida de peso, astenia), poliadenopatías, hepatoesplenomegalia, anemia, afección pulmonar bilateral (similar a la hallada en la TBC miliar), lesiones cutáneas múltiples (de tipo acneiforme, papuloulceradas, púrpuricas o papulovesiculosas, nódulos ulcerados), siendo las lesiones osteoarticulares de aparición excepcional, pero en los casos más severos puede llegar a producir un cuadro de osteomielitis crónica con lesiones osteolíticas. Puede haber compromiso vertebral en forma de espondilitis, pero es aún más raro. Sin tratamiento conduce a la muerte por diseminación en pocos meses. Tanto por la clínica como por el tiempo de evolución lo considero un diagnóstico alejado.
Dentro de las etiologías neoplásicas debemos considerar principalmente los linfomas, las metástasis de tumores sólidos y en último lugar la histiocitosis maligna que es una patología muy poco frecuente.
La presentación clínica de los linfomas no Hodgkin (LNH) varía ampliamente dependiendo del tipo histológico y de los órganos afectados. Algunos presentan un curso indolente con desarrollo de linfadenopatías, mientras otros son altamente agresivos llevando a la muerte en pocas semanas si no se tratan.
Los linfomas agresivos se presentan más comúnmente con una masa de rápido crecimiento, síntomas B (fiebre, sudoración nocturna, pérdida de peso), y/o niveles elevados de LDH y ácido úrico. Estos comprenden al linfoma difuso de células B, linfoma de Burkitt, linfoma de células T.
Los linfomas de curso indolente suelen presentarse con linfadenopatías de crecimiento lento, hepatoesplenomegalia o citopenias. En este grupo: linfoma folicular, linfoma linfocítico de células pequeñas.
Presentaciones menos frecuentes incluyen rash cutáneo, prurito, ascitis y derrames. Aproximadamente 50% de los pacientes desarrollarán enfermedad extranodal en el curso de la enfermdad, mientras que 10-35% presentarán compromiso extranodal al momento del diagnóstico.
La piel es el segundo sitio más común de afectación extranodal. Los pacientes con linfoma de células T pueden desarrollar lesiones cutáneas de tipo paniculitis, indoloras que aparecen principalmente a nivel de miembros inferiores y superiores, tronco y cara. En el 80% de los casos las lesiones suelen ser múltiples.
La afectación ósea es usualmente manifestación de una enfermedad diseminada, pudiendo presentarse incluso como una fractura patológica.
El linfoma de Hogdkin (LH) presenta una distribución etaria bimodal con un 1er pico de incidencia entre los 20-30 años y el 2do luego de los 50.
Las manifestaciones clínicas son inespecíficas pudiendo confundirse inicialmente con cuadros infecciosos. Afecta más comúnmente los ganglios cervicales (60-80%), aunque también puede presentarse con adenopatías mediastinales (60-70%) y retroperitoneales (25%) o con el hallazgo de una masa mediastinal en la Rx tórax realizada por otro motivo. La afectación extranodal es rara (<1%; más frecuente en LNH), siendo el sitio más frecuentemente comprometido el tracto gastrointestinal. El derrame pleural es poco frecuente, excepto en pacientes con una masa mediastinal de gran tamaño.
La presencia de síntomas B se observa usualmente en estadíos avanzados de la enfermedad.
Una variedad de lesiones cutáneas se ha asociado con el LH, incluyendo ictiosis, urticaria, eritema multiforme, eritema nodoso, lesiones necrotizantes, hiperpigmentación e infiltración cutánea.
Puede producir múltiples alteraciones analíticas, entre las que se describen: hipercalcemia, anemia, eosinofilia, leucocitosis, trombocitosis e hipoalbuminemia.
El síndrome de compresión medular se ha descripto como una forma de presentación, pero es poco frecuente.
Tipícamente es una patología sistémica que se presenta con compromiso óseo en un 10-20% de casos durante la evolución de la misma, correspondiendo a un estadio IV en la clasificación de Ann Arbor.
Los linfomas óseos primarios son tumores raros, representando entre el 3-5% de todos los tumores óseos primarios, con mayor incidencia en la adolescencia temprana y leve predilección por el sexo masculino.
Representa menos del 2% de todos los linfomas en adultos, la mayoría de ellos son LNH y de linaje B (70%). El diagnóstico es más frecuente en pacientes mayores de 30 años (92%).
Sus manifestaciones clínicas son inespecíficas, siendo el dolor óseo que no mejora con el descanso el síntoma más frecuente. Otros signos y síntomas que pueden presentarse en el momento del diagnóstico son: masa palpable en el 50% de los casos, fracturas patológicas, compresión medular y síntomas B (fiebre, pérdida de peso, sudoración nocturna, etc). La poca especificidad de los síntomas puede conllevar un retraso significativo en el diagnóstico.
La afectación puede ser uni o multifocal con predilección por el esqueleto axial. Los huesos afectados con más frecuencia son fémur, tibia, pelvis y columna vertebral.
Los principales diagnósticos diferenciales son el osteosarcoma, sarcoma de Ewing, histiocitos de Langerhans, linfoma con afectación ósea y osteomielitis y en pacientes mayores, las metástasis óseas.
Creo que el linfoma es un diagnóstico probable en el caso en discusión, sobre todo de tipo no Hodgkin con afectación ósea de forma secundaria.
La histiocitosis de Langerhans (HCL) es una condición rara que se caracteriza por la proliferación clonal anormal de la célula de Langerhans. Son células dendríticas intraepidérmicas cuyo papel principal es la presentación de antígeno a las células T efectoras.
Su etiología es aún desconocida, aunque muchos autores proponen que se debe a una disfunción del sistema inmune.
Se puede presentar con afectación local o sistémica.
La HCL puede presentarse en cualquier edad, desde el periodo neonatal a la edad adulta. El pico de incidencia está entre el primero a tercer año de vida. La incidencia estimada anual es de 4 casos por millón. Los hombres son afectados con mayor frecuencia que las mujeres, en una relación 2:1.
El espectro clínico general de la HCL incluye compromiso de la piel y anexos, hueso, médula ósea, hígado, bazo, ganglios linfáticos, pulmón, tracto gastrointestinal inferior, sistema endocrino y sistema nervioso central.
Las lesiones óseas son la manifestación más frecuente de la HCL (80%) y se asocian con buen pronóstico. Con frecuencia se trata de lesiones asintomáticas; sin embargo, puede presentarse con dolor local, deformidad ósea o fracturas patológicas.
Los huesos afectados con mayor frecuencia son el cráneo, los huesos largos y los huesos planos.
La incidencia de compromiso espinal varía entre 6.5-25% de los casos con afectación ósea y suele tratarse de una lesión única. En la columna, la HCL afecta principalmente los cuerpos vertebrales, con predilección por la columna dorsal (54%).
Las lesiones cutáneas son comunes (40% de las HCL) y frecuentemente la manifestación inicial de la enfermedad. Las localizaciones más frecuentes son el cuero cabelludo, el tronco y las áreas flexurales. Las lesiones iniciales consisten en pápulas rosadas o cafés que con el tiempo pueden llegar a ser confluentes, descamativas y costrosas.
Los ganglios linfáticos pueden estar comprometidos en un pequeño número de pacientes con HCL. Su presencia puede ser parte de una enfermedad diseminada o estar asociado sólo con enfermedad local de la piel o el hueso adyacente. Los ganglios linfáticos cervicales son los más afectados.
El compromiso de la médula ósea es raro y ocurre de forma tardía en el curso de la enfermedad. Su disfunción puede llevar a anemia, leucopenia, trombocitopenia o pancitopenia, principalmente en pacientes con enfermedad diseminada.
El compromiso pulmonar es frecuente, especialmente en pacientes con enfermedad multisistémica. Los hallazgos radiológicos pueden mostrar micronódulos difusos y tardíamente un patrón reticulonodular o miliar intersticial. De manera tardía pueden ocurrir cambios enfisematosos con fibrosis intersticial difusa.
Si bien el cuadro clínico parece ser similar al de nuestro paciente, lo que lo hace un diagnóstico posible, no debemos olvidar la rareza de esta enfermedad y siempre considerar inicialmente las patologías de presentación más habitual.
Dentro de las misceláneas haré mención de 4 patologías, siempre recordando la baja frecuencia con la que se presentan.
La sarcoidosis es un desorden sistémico caracterizado por la formación de granulomas no caseificantes en los órganos comprometidos. La prevalencia es de 1-40 casos/100000 habitantes, afectando habitualmente adultos menores de 40 años de ambos sexos.
Puede ser autolimitada o crónica con exacerbaciones y remisiones. Puede afectarse prácticamente cualquier órgano. Los pulmones (90%) y los ganglios torácicos son los más comúnmente comprometidos.
La mayoría de pacientes presentan síntomas sistémicos como fatiga, anorexia, pérdida de peso y fiebre.
El compromiso pulmonar ocurre en más del 90% de los pacientes con sarcoidosis, hallándose en Rx tórax adenopatías hiliares bilaterales e infiltrados intersticiales o alveolares. La afección pleural es inusual (<5%) pero puede presentarse con derrame pleural linfocítico, quilotórax, neumotórax y hemotórax.
En todos los casos sospechosos debe confirmarse el diagnóstico mediante la realización de biopsia para descartar infección y malignidad. Es un diagnóstico de exclusión.
Aproximadamente 25% de los pacientes tendrán una o más manifestaciones cutáneas, entre las que se describen el eritema nodoso, nódulos subcutáneos, máculas, pápulas y lesiones cicatrizales.
No se encuentran en la literatura casos descriptos con compromiso óseo y espinal.
La enfermedad de Castleman es un desorden muy poco frecuente caracterizado por la proliferación benigna de tejido linfoide en relación a infección crónica por HHV-8 y HIV. Se describen 2 formas de presentación: localizada con compromiso de un único ganglio linfático, y la sistémica con desarrollo de adenopatías generalizadas y curso clínico más agresivo. Se acompaña de afección sistémica (fiebre, sudoración nocturna) y hepatoesplenomegalia. Se ha descripto su asociación con neoplasias, particularmente con el sarcoma de Kaposi y los linfomas.
La enfermedad de Kimura es un desorden inflamatorio crónico raro de etiología desconocida que afecta principalmente a la población asiática. Usualmente se presenta entre los 20-30 años, predominando en el sexo masculino. Se cree que el curso sería benigno. Se caracteriza por presentarse con eosinofilia y niveles elevados de IgE. La forma de presentación más común es con linfadenopatías cervicales, indoloras o masas subcutáneas sobre todo a nivel de cabeza y cuello.
Le enfermedad de Gaucher es un trastorno autosómico recesivo causado por un defecto en la glucosidasa beta ácida. Las variantes de la enfermedad se clasifican de acuerdo a la presencia o ausencia y gravedad de la afección neuropática.
Le enfermedad de Gaucher tipo 1 es un trastorno no neuropático que puede manifestarse en la infancia o en la edad adulta. Tiene una presentación típicamente bimodal con picos de incidencia máxima entre 10-15 años y alrededor de los 25. Los pacientes más jóvenes tienen hepatoesplenomegalia y citopenias, mientras que en el grupo de mayor edad es mayor la tendencia a la enfermedad ósea crónica. Todos los pacientes sintomáticos presentan hepatoesplenomegalia leve o masiva.
La remodelación del hueso está alterada; la pérdida del calcio óseo total da por resultado osteopenia, osteoporosis y fracturas vertebrales por compresión que lesionan la médula espinal.
Ante el antecedente que refiere el paciente me pregunto si nos encontramos ante una fractura patológica con posterior desarrollo de una tumoración o si la alteración ósea es secundaria a un proceso invasivo. En cualquiera de los 2 casos, esto nos lleva a pensar en los tumores óseos y de partes blandas como probables etiologías.
Debe sospecharse la presencia de una fractura patológica en un paciente joven cuando la misma se asocia a un trauma mínimo, cuando la localización es inusual o cuando se observan alteraciones óseas en las radiografías. En los adolescentes y adultos jóvenes, la mayoría de fracturas patológicas se deben a tumores óseos benignos, alteraciones metabólicas o infecciones. De cualquier manera, la presencia de un proceso maligno siempre debe sospecharse porque también puede ser una de las causas.
Las fracturas patológicas son la forma de presentación de 5-10% de los procesos malignos primarios del hueso. La falla en el reconocimiento de estas fracturas lleva a un tratamiento inadecuado y a largo plazo a un potencial peor pronóstico.

La clavícula es un sitio en el que raramente asientan tumores óseos primarios (incidencia: 0.45-1%), por lo que el diagnóstico puede retrasarse por el bajo nivel de sospecha.
El osteosarcoma y el sarcoma de Ewing son los tumores malignos óseos más frecuentes en los jóvenes, mientras que las metástasis de tumores sólidos son la causa más frecuente de una lesión tumoral ósea en la población general.
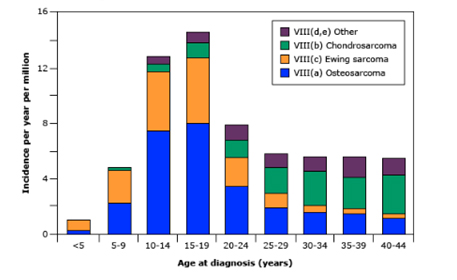
El osteosarcoma es más frecuente en varones, en la segunda década de la vida. Es un tumor poco común, pero a pesar de esto es la neoplasia maligna ósea primaria más frecuente en niños y adolescentes. La mayoría de pacientes con osteosarcoma se presentan con dolor localizado de meses de duración, el que comienza habitualmente luego de una injuria. La afección sistémica (fiebre, pérdida de peso) generalmente está ausente. El principal hallazgo en el examen físico es la presencia de una masa de tejidos blandos. Suelen localizarse en los huesos largos (fémur, tibia, húmero).
El laboratorio habitualmente no presenta alteraciones, excepto por elevaciones de la fosfatasa alcalina (40% casos), LDH (30%) y VES. La elevación de la LDH se asocia con un peor pronóstico.
Las metástasis más frecuentes son a nivel de pulmón (90%), pleura y huesos (10%).
El sarcoma de Ewing predomina en las dos primeras décadas de la vida y en el sexo masculino, afectando principalmente a los huesos largos (pelvis y esqueleto axial), aunque puede desarrollarse prácticamente en cualquier hueso o tejido blando. El 50% de los mismos asientan de forma primaria en la pared torácica, afectando sobre todo las costillas. Clínicamente se presenta con dolor localizado de semanas o meses de duración. Un trauma, incluso menor, puede ser el evento desencadenante que pone la atención sobre la lesión. Al examen físico puede evidenciarse una masa de tejidos blandos que suele estar adherida al plano óseo.
La presencia de síntomas constitucionales como fiebre, fatiga, pérdida de peso o anemia se encuentra en 10-20% de los pacientes al momento del diagnóstico.
Los niveles altos de LDH se asocian con enfermedad metastásica.
Produce metástasis de forma temprana en pulmones y otros huesos, hallándose hasta en un 25% de pacientes al momento del diagnóstico. La médula espinal es el sitio más frecuentemente comprometido a nivel óseo de forma secundaria, pudiendo presentarse con síntomas compatibles con un síndrome de compresión medular.
El condrosarcoma es un tumor de crecimiento lento y que provoca dolor y tumefacción local, predomina en varones de edad avanzada. Las lesiones se presentan sobre todo a nivel del esqueleto axial y en los huesos largos. Las metástasis son poco frecuentes.
Ante la presencia de una importante tumoración a nivel de la pared torácica no debemos olvidar otros importantes diagnósticos diferenciales como son el rabdomiosarcoma y el tumor neuroectodérmico primitivo (tumor de Astkin) que son las neoplasias malignas de tejidos blandos más frecuentes durante la infancia.
El tumor neuroectodérmico primitivo (PNET) es una neoplasia extremadamente agresiva que afecta principalmente a niños, cerca del 80% de los pacientes son menores de 20 años y es rara por encima de los 30. Pertenece a la familia de los tumores de Ewing. Puede manifestarse como una tumoración a nivel de la pared torácica con destrucción de la costilla adyacente, engrosamiento pleural o derrame e invasión pulmonar local.
Los rabdomiosarcomas son neoplasias torácicas que pueden afectar pulmones, bronquios, mediastino, corazón y pared torácica. Se presentan durante la infancia o entre los 50-70 años predominando en el sexo masculino. A diferencia del sarcoma de Ewing y el PNET suele ser inusual el compromiso costal.
Por lo expuesto anteriormente, considero que entre los tumores óseos y de partes blandas primarias, el cuadro clínico de nuestro paciente sería más compatible con la presentación de un sarcoma de Ewing.
En cuanto a las alteraciones del laboratorio, me pregunto si las mismas pueden darnos una pista sobre la probable etiología del cuadro que presenta el paciente. Recordemos que el mismo presenta: anemia de enfermedades crónicas, eosinofilia, trombocitosis, VES acelerada, aumento de LDH, hiperuricemia e hiperfosfatemia.
Anemia de enfermedades crónicas. Clásicamente considerada como aquella que acompaña a procesos inflamatorios, infecciosos y neoplásicos; es un dato inespecífico.
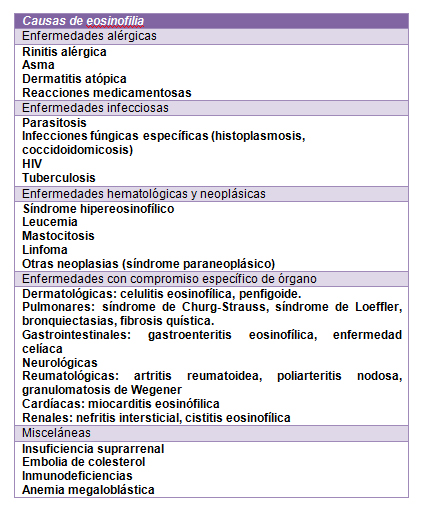
Eosinofilia. Cuando la cifra de eosinófilos supera los 500/ml. Existen numerosas causas que pueden provocar la aparición de eosinofilia, asociándose de forma más frecuente a la existencia de procesos alérgicos, infecciosos o neoplásicos, aunque también puede evidenciarse ante la ingesta de determinados fármacos o en el contexto de colagenopatías.
Trombocitosis. Puede ser secundaria (reactiva) o primaria. Entre las posibles causas de una trombocitosis secundaria se incluyen la hemorragia reciente, anemia ferropénica, infecciones agudas, postoperatorio de cirugías mayores, neoplasias (especialmente enfermedad de Hogdkin, aunque también puede acompañar a síndromes mieloproliferativos crónicos como la LMC, policitemia vera o mielofibrosis primaria), enfermedades autoinmunes (EII, AR) como durante la recuperación tras tratamientos con radioterapia o quimioterapia.
Incremento de LDH. Las causas de su elevación son múltiples: cardiovasculares, hepáticas, hematológicas, musculares, pulmonares, neoplásicas, nefrológicas y otras. Es una herramienta poco específica y que no es de utilidad en el contexto del paciente.
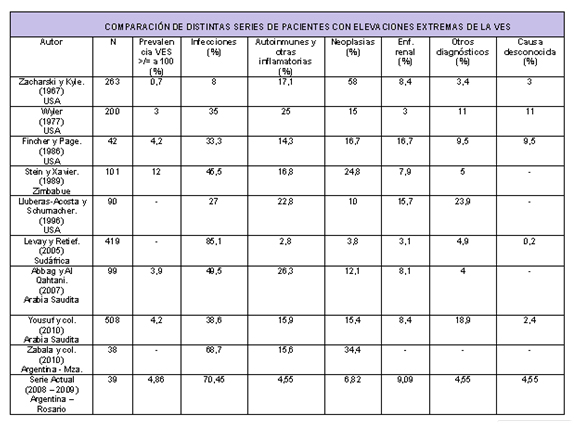
VES acelerada. A pesar de que es una determinación inespecífica, una elevación extrema (>100 mm en 1er hora) está asociada en más del 95% de los casos con una enfermedad seria subyacente. En la mayoría de las series las infecciones constituyen las causas predominantes, seguidas por las enfermedades del colágeno y los tumores metastásicos.
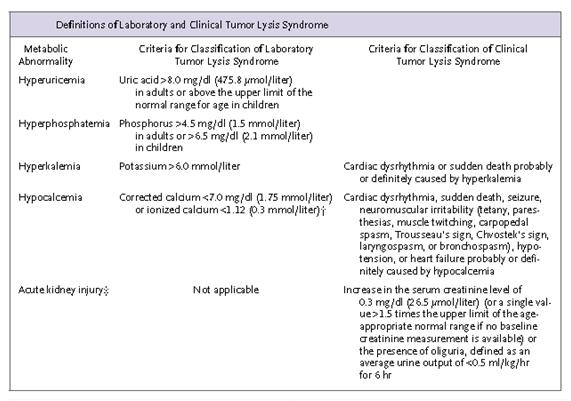
Hiperuricemia e hiperfosfatemia. Ante la presencia simultánea en el laboratorio de hiperuricemia e hiperfosfatemia que se desarrollaron tras 24 hs de tratamiento con esteroides a dosis altas, me pregunto si nos encontramos ante un síndrome de lisis tumoral.
El síndrome de lisis tumoral (SLT) analítico se define por la presencia simultánea de 2 o más alteraciones metabólicas (hiperuricemia, hiperkalemia, hiperfosfatemia e hipocalcemia) 3 días previo o hasta 7 días del inicio de la terapéutica. El SLT clínico se presenta cuando las alteraciones analíticas se acompañan por un cuadro de insuficiencia renal, arritmia cardíaca, convulsiones o falla multiorgánica.
El mismo ocurre cuando las células tumorales vuelcan su contenido en el torrente sanguíneo, ya sea espontáneamente o en respuesta al tratamiento.
Las neoplasias que conllevan alto riesgo de lisis incluyen los linfomas de alto grado, leucemias agudas y otras neoplasias de rápida proliferación.
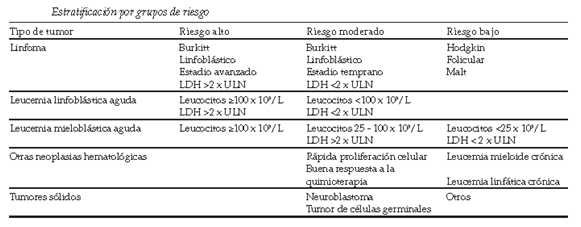
El síndrome de compresión medular (SCM) es una complicación grave que ocurre en el 5-10% de pacientes con cáncer. Su incidencia varía con el tipo de tumor: los cánceres de próstata, mama y pulmón representan el 45-60% de todos los SCM, los mielomas múltiples y linfomas el 5-10% de los SCM y con menor frecuencia aparece también en otros tumores que presenten diseminación ósea durante su evolución. Hasta un 30% de los SCM son la primera manifestación de una neoplasia aún no diagnosticada.
Es ampliamente aceptado que la compresión medular neoplásica es una emergencia médica, requiriendo un diagnóstico y tratamiento precoz debido a que los retrasos pueden resultar en pérdidas funcionales irreversibles, lo cual condiciona un empeoramiento notable en la calidad de vida. El factor pronóstico más importante es la función neurológica previa a la instauración de la terapéutica.
Las demoras entre el comienzo de los síntomas y el tratamiento suele ser mayor en aquellos pacientes que no presentan diagnóstico previo de neoplasia.
La RMI, con una sensibilidad y especificidad del 93% y 97% respectivamente, es el estudio de imágenes de elección.
El SCM afecta a la columna dorsal entre el 60 y 80% de las veces, en el 15-30% de las ocasiones asienta sobre la columna lumbar y menos del 10% de las veces afecta de manera exclusiva a la columna cervical. Más de la mitad de los pacientes presentan al diagnóstico afectación simultánea de más de un área de la columna vertebral.

Para concluir nos encontramos ante un paciente varón, joven sin antecedentes de jerarquía que consulta por síntomas constitucionales, presentando al examen físico una tumoración clavicular, múltiples nódulos subcutáneos y una paraparesia crural; evidenciándose en el laboratorio una elevación extrema de la VES, hiperuricemia e hiperfosfatemia, por lo que me pregunto si nos encontramos ante un síndrome de lisis tumoral. Presenta además en los estudios por imágenes realizados un derrame pleural unilateral y una masa con compromiso medular. Creo que nos encontramos ante una patología de marcada agresividad, siendo las probables etiologías infecciosas versus neoplásicas. Sin embargo, por todo lo expuesto considero que las neoplasias son el diagnóstico más probable y dentro de estas en primer lugar los linfomas y posteriormente los sarcomas.
Bibliografía
- Mathurin S, Agüero A et al. Enfoque clínico del paciente con adenopatías. 2008 Clínica-UNR.org Publicación digital de la 1ra Cátedra de Clínica Médica y Terapéutica y la Carrera de Posgrado de especialización en Clínica Médica Facultad de Ciencias Médicas - Universidad Nacional de Rosario.
- Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, et al. (2012) Optimal Therapy for Adults with Langerhans Cell Histiocytosis Bone Lesions. PLoS ONE 7(8): e43257.
- Gomez-Ramirez J, Posada M et al. Castleman’s Disease: A Case Report of the Unicentric Type. Hindawi Publishing Corporation, Case Reports in Surgery Volume 2012.
- Van den Akker et al.: An osseous lesion in a 10-yearold boy with Hodgkin’s lymphoma: a case report. Journal of Medical Case Reports 2011 5:511.
- Li et al.: Unusual primary osseous Hodgkin lymphoma in rib with associated soft tissue mass: a case report and review of literature. Diagnostic Pathology 2012 7:64.
- De Mattos C, Binitie O, Dormans J. Pathological fractures in children. Bone Joint Res 2012;1:272–80.
- Tan J, Schlicht P et al. Multidisciplinary approach to diagnosis and management of
osteosarcoma – a review of the St Vincent's Hospital experience. International Seminars in Surgical Oncology 2006, 3:38
- Nam S, Sungjun K et al.Imaging of Primary Chest Wall Tumors with Radiologic-Pathologic Correlation. RadioGraphics 2011; 31:749–770
- Krassas A, Mallios D et al. Primitive Neuroectodermal Tumor of the Thoracic Wall in a 48-Year-Old Man. Asian Cardiovascular and Thoracic Annals 2010 18: 285.
- Gladish G, Sabloff et al. Primary Thoracic Sarcomas. RadioGraphics 2002; 22:621–637
- Parodi R, Chorzepa G. Eosinofilia. En: Greca A, Gallo R, Parodi R. Medicina Ambulatorio. 1º edición. Corpus, Rosario 2007; 403-409.
- Allan L, Baker L et al. Suspected malignant cord compression – improving time to diagnosis via a ‘hotline’: a prospective audit. British Journal of Cancer (2009) 100, 1867–1872.
- Husband D. Malignant spinal cord compression: prospective study of delays in referral and treatment. BMJ 1998;317:18–21.
- Hernanz de Lucas R, Montero L et al. Síndrome de compresión medular en urgencias: utilización de un algoritmo diagnóstico-terapéutico. Emergencias 2010; 22: 120-124.
- Romero P, Manterola A. Compresión medular. An. Sist. Sanit. Navar. 2004; 27 (Supl. 3): 155-162.
- Luo CC. Spinal cord compression secondary to metastatic non-Hodgkin’s lymphoma: a case report. Arch Phys Med Rehabil 2005;86:332-4.
|
|
|
Discusión |
|
La discusión de este seminario
corresponde al 23 de Mayo de 2013, a
cargo de Dra. Gisela Goldberg |
|
| |
|
|
| |
|
|
|
|
|

